Zespół Pfeiffera
Zespół Pfeiffera to rzadkie zaburzenie cechujące się przedwczesnym zrastaniem szwów czaszki (craniosynostosis), szerokimi krzywymi kciukami i paluchami, oraz częściowo zrośniętymi palcami u rąk i nóg (syndaktylia). Choroba dotyka 1 na 100000 osób.
Objawy
- Craniosynostosis (kraniosynostoza, czyli przedwczesne zarośnięcie szwów czaszki) oraz krótkie, grube kciuki i paluchy. Kraniosynostoza to proces przedwczesnej fuzji stawów włóknistych kości czaszki. U zdrowego dziecka czaszka się powiększa wraz z rosnącym mózgiem. U dziecka z kraniosynostozą jeden lub dwa szwy zarastają zbyt wcześnie powodując nieprawidłowy, asymetryczny rozrost czaszki i twarzy. Gdy proces craniosynostosis jest zaawansowany, tzn. dotyczy wielu szwów, może dojść do wzrostu ciśnienia wewnątrzczaszkowego lub zahamowania rozwoju mózgu. Objawiać się to może opóźnieniem rozwoju umysłowego, napadami drgawek lub ślepotą. Szwy najczęściej dotknięte w przebiegu zespołu Pfeiffera to szew wieńcowy, węgłowy i strzałkowy
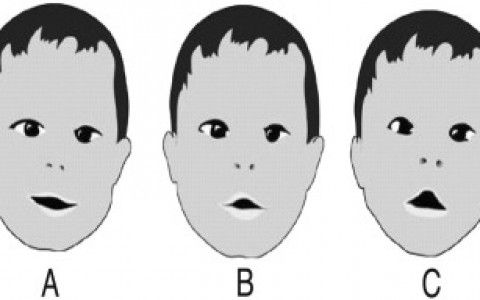
- Nieproporcjonalnie szeroka głowa z wysokim czołem oraz cofnięty, zapadnięty środek twarzy (obszar pomiędzy oczodołami a górną szczęką). Nos jest często mały z niskim grzbietem. Oczy mogą być szeroko rozstawione (hiperteloryzm) i wytrzeszczone (proptotyczne) z powodu płytkich oczodołów
- Upośledzenie słuchu spowodowane niezwykle małym przewodem słuchowym i uchem środkowym (dotyczy ok. 50% dzieci z zespołem Pfeiffera)
- Problemy stomatologiczne
- Zaburzenia wzorku spowodowane nieprawidłowym położeniem oczu i zwiększonym ciśnieniem środczaszkowym spowodowanym przedwczesnym zrostem szwów czaszki
Przyczyny
Zespół Pfeiffera jest wywołany mutacją genu sterujące receptorem czynnika wzrostu fibroblastów (FGFR = fibroblast growth factor receptor, 1 lub 2). Geny FGFR grają ważną rolę w stymulowaniu komórki do podziału lub do dojrzewania. Zaburzone funkcjonowanie tego genu powoduje przedwczesne zrastanie się kości czaszki lub palców rąk i nóg. Niektóre badania wskazują, że zespół ten występuje częściej u dzieci, których ojcowie są relatywnie starsi.
Zespół Pfeiffera obejmuje trzy podtypy zależnie od natężenia objawów
Typ 1: Osoby z tym typem cechują: przedwcześnie zarośnięte szwy czaszki, wklęsłe kości policzkowe, zniekształcone palce. Rozwój neurologiczny i umysłowy jest zwykle normalny. Może wstępować wodogłowie i upośledzenie słuchu.
Typ 2: Pacjenci z tym typem mają czaszkę zdeformowaną na kształt „koniczyny”, co jest wynikiem rozległego przedwczesnego zrostu szwów czaszki. Towarzyszy wytrzeszcz, zniekształcenia palców, zrosty w stawach łokciowych i kolanowych (ankylosis). Zniekształcenie czaszki może powodować ograniczenie rozwoju mózgu i upośledzenie umysłowe. Proptoza oczu może być przyczyną ciężkich zaburzeń widzenia. Wiele osób z tym typem jest opóźnionych w rozwoju fizycznym i umysłowym.
Typ 3: Chorzy z tą odmianą zespołu Pfeiffera mają objawy podobne do typu 2, lecz ich czaszka nie jest zdeformowana na kształt „koniczyny”.
Diagnoza i leczenie
Diagnoza zostaje oparta na obecności przedwczesnej fuzji kości czaszki i szerokich, krótkich kciukach i paluchach. Inne zespoły, które należy brać pod uwagę w diagnostyce różnicowej to: zespół Aperta, zespół Crouzona, zespół Saethre-Chotzen i zespół Jackson-Weiss.
Trudno jest rozpoznać zespół Pfeiffera prenatalnie za pomocą ultrasonografii, gdyż cechy kliniczne przybierają bardzo różne formy.
Leczenie dziecka z zespołem Pfeiffera zaczyna się w momencie narodzin od dokładnej diagnozy, identyfikacji potrzeb dziecka. Terapia może obejmować różne zabiegi chirurgiczne, przeprowadzone przez wielodyscyplinarny zespół specjalizujący się w chorobach twarzoczaszki. W skład takiego zespołu wchodzą neurochirurg, chirurg plastyczny, dentysta, ortodonta, audiolog, logopeda, otorynolaryngolog, genetyk i pediatra.
Wczesna operacja mająca na celu uwolnienie przedwcześnie zarośniętych szwów czaszki może być zalecona w pierwszym roku życia. Uwolnienie szwów pozwoli mózgowi na normalną ekspansję, a czaszce na prawidłowy wzrost. W trakcie tej samej operacji można powiększyć oczodoły poprawiając widzenie. Środek twarzy można chirurgicznie modelować w późniejszym wieku w celu poprawy wyglądu pacjenta, zwiększenia pojemności oczodołów i ustalenia bardziej prawidłowej relacji pomiędzy górną a dolną szczęką.
Inne terapie
- testy audiologiczne, aby ustalić, czy potrzebny jest zabieg chirurgiczny ucha celem zachowania słuchu
- konsultacja stomatologiczna w drugim roku życia
- chirurgia ręki w celu uwolnienia zrośniętych palców